Nextflow (& some other workflow systems)
Rad Suchecki
Monday, 25 June, 2018

Let others (and yourself!)
- reliably re-run your analyses
- trace back origins of results
triggers
- new data (e.g. additional samples)
- updated software
- different compute environment (cloud?)
- errors found
- new ideas
A workflow management system might help…
Aims & considerations
- turn-key reproducibility
- re-usability

Workflow systems
- 214 systems and counting
- from
maketo Galaxy - see also this list
- from
- active development?
- (community) support?
- flexibility and ease of development
- learning curve?
- abstraction level - just right ;-)
- …
Non-GUI, bioinf-flavoured and… lively
- Snakemake: a pythonic workflow system
- follows
makephilosophy - rules and patterns that depend on input/output file names
- explicit, precomputed DAG
- follows
- Nextflow
- dataflow programming model - think Unix pipes
- conditional paths which may depend on intermediate results
- DAG not known in advance
- Comparisons
- 2018 https://vatlab.github.io/blog/post/comparison/ (by SoS authors)
- 2017 https://www.nature.com/articles/nbt.3820/tables/1 (by NF authors)
- 2016 https://jmazz.me/blog/NGS-Workflows (blog)
- 2016 https://doi.org/10.1093/bib/bbw020 (review, not quite right about NF)
Nextflow
- Designed for
- specific use case: seamless scalability of existing tools and scripts
- for bioinformaticians familiar with programming
Features
- Separation of pipeline logic from compute environment definition(s)
- Syntax is a superset of Groovy, but polyglot
- easily mix scripting languages
- Multiple executors
- SGE, LSF, SLURM, PBS/Torque, NQSII HTCondor, Ignite, Kubernets, AWS Batch
- NEW! Support for Conda/Bioconda
More about Nextflow
Processes (tasks) executed in separate work directories
- safe and lock-free parallelization
- easy clean-up, no issue of partial results following an error
Channels
- facilitate data flow between processes
Getting started
Required
- POSIX compatible system (Linux, Solaris, OS X, etc)
- Java 8
Install
curl -s https://get.nextflow.io | bashSoftware you want to run
- Available on PATH or under
bin/ - via Docker
- via Singularity
- via Conda
- via Modules
Hello world syntax
#!/usr/bin/env nextflow
echo true
cheers = Channel.from 'Bonjour', 'Ciao', 'Hello', 'Hola'
process sayHello {
input:
val x from cheers
script:
"""
echo '$x world!'
"""
}Hello world shared pipelines
N E X T F L O W ~ version 0.30.1
Pulling nextflow-io/hello ...
downloaded from https://github.com/nextflow-io/hello.git
Launching `nextflow-io/hello` [friendly_roentgen] - revision: c9b0ec7286 [master]
[warm up] executor > local
[5c/104ae2] Submitted process > sayHello (2)
[d7/11b28c] Submitted process > sayHello (1)
[4e/e5e39c] Submitted process > sayHello (3)
Ciao world!
Bonjour world!
[15/9f213a] Submitted process > sayHello (4)
Hello world!
Hola world! project name: nextflow-io/hello
repository : https://github.com/nextflow-io/hello
local path : /home/rad/.nextflow/assets/nextflow-io/hello
main script : main.nf
revisions :
* master (default)
mybranch
v1.1 [t]
v1.2 [t]Run specific revision (tag/branch/commit SHA hash)
N E X T F L O W ~ version 0.30.1
Launching `nextflow-io/hello` [confident_kalam] - revision: 451ebd9dcb56045d80963945305811aa65f413d0
[warm up] executor > local
[e0/bbb32c] Submitted process > sayHello (2)
[c0/c7c8ef] Submitted process > sayHello (1)
[f9/e08c9a] Submitted process > sayHello (3)
Bojour world!
Ciao world!
[9a/195a35] Submitted process > sayHello (4)
Hello world!
Hola world!Something a bit more useful

N E X T F L O W ~ version 0.30.1
Launching `nextflow-io/blast-example` [furious_lovelace] - revision: 34a29e1cdc [master]
[warm up] executor > local
[5b/2f03b9] Submitted process > blast (1)
[de/c4a679] Submitted process > extract (1)
matching sequences:
>lcl|1ABO:B unnamed protein product
MNDPNLFVALYDFVASGDNTLSITKGEKLRVLGYNHNGEWCEAQTKNGQGWVPSNYITPVNS
>lcl|1ABO:A unnamed protein product
MNDPNLFVALYDFVASGDNTLSITKGEKLRVLGYNHNGEWCEAQTKNGQGWVPSNYITPVNS
>lcl|1YCS:B unnamed protein product
PEITGQVSLPPGKRTNLRKTGSERIAHGMRVKFNPLPLALLLDSSLEGEFDLVQRIIYEVDDPSLPNDEGITALHNAVCA
GHTEIVKFLVQFGVNVNAADSDGWTPLHCAASCNNVQVCKFLVESGAAVFAMTYSDMQTAADKCEEMEEGYTQCSQFLYG
VQEKMGIMNKGVIYALWDYEPQNDDELPMKEGDCMTIIHREDEDEIEWWWARLNDKEGYVPRNLLGLYPRIKPRQRSLA
>lcl|1IHD:C unnamed protein product
LPNITILATGGTIAGGGDSATKSNYTVGKVGVENLVNAVPQLKDIANVKGEQVVNIGSQDMNDNVWLTLAKKINTDCDKTCompute profiles
- Implemented/tested with my pipelines
standard(i.e. local/interactive assuming all software available)slurmmodulesdockersingularityec2(including auto-scaling)conda(missing a bioconda recipe for biokanga)
# some of the profiles can, and others are intended to be used in concert, e.g.
nextflow run csiro-crop-informatics/reproducible_poc -r develop -profile slurm,modules
# or
nextflow run csiro-crop-informatics/reproducible_poc -r develop -profile slurm,singularity,singularitymodule- Next:
k8s,awsbatch, …
Modules
- Required modules are defined separately for each process (
withName) or for multiple processes (withLabel)
process {
withName: fastQC {
module = 'fastqc/0.11.5'
}
withName: multiQC {
module = 'python/2.7.13'
}
withName: hisat2Index {
module = 'hisat/2.0.5'
}
withName: hisat2Align {
module = ['hisat/2.0.5','samtools/1.7.0']
}
withLabel: biokanga {
module = 'biokanga/4.3.9'
}
withLabel: samtools {
module = 'samtools/1.7.0'
}
}- Note! These are compute environment specific
Containers
- Required containers are defined separately for each process (
withName) or for multiple processes (withLabel) - Available on docker hub
- Used both by
dockerandsingularity
- Used both by
process {
withName: extractStatsFromBAMs {
container = 'genomicpariscentre/samtools:1.4.1'
}
withName: fastQC {
container = 'genomicpariscentre/fastqc:0.11.5'
}
withName: multiQC {
container = 'ewels/multiqc:v1.5'
}
withName: fasta2mockFASTQ {
container = 'rsuchecki/pigz:2.3.4'
}
withLabel: biokanga {
container = 'rsuchecki/biokanga:4.3.9'
}
withLabel: hisat2 {
container = 'cyverse/hisat2:2.0.5'
}
}Workflow introspection
NF Resources
Future?
- http://nf-co.re/
- https://github.com/assemblerflow/flowcraft (Or a step too far?)
According to Nextflow authors
- The dataflow model is superior to alternative solutions based on a Make-like approach, such as Snakemake, in which computation involves the pre-estimation of all computational dependencies, starting from the expected results up until the input raw data. A Make-like procedure requires a directed acyclic graph (DAG), whose storage requirement is a limiting factor for very large computations. In contrast, as the top to bottom processing model used by Nextflow follows the natural flow of data analysis, it does not require a DAG. Instead, the graph it traverses is merely incidental and does not need to be pre-computed or even stored, thereby ensuring high scalability.
This slide is intentionally left blank
Pipeline run (I)
N E X T F L O W ~ version 0.30.1
Launching `csiro-crop-informatics/reproducible_poc` [infallible_brattain] - revision: e957f4b275 [develop]
NOTE: Your local project version looks outdated - a different revision is available in the remote repository [5b495fe02f]
[warm up] executor > local
[12/5181e6] Submitted process > fetchRef (A_thaliana)
[c2/8d5f02] Submitted process > fetchReads
[ba/6e35da] Submitted process > hisat2Index (A_thaliana)
[da/71e1b9] Submitted process > kangaIndex (A_thaliana)
[e6/2bf4e9] Submitted process > fastQC ([name:real, nreads:10k, seqerr:unk, rep:na, format:fq])
[29/8e7364] Submitted process > hisat2Align ([name:real, nreads:10k, seqerr:unk, rep:na, format:fq, ref:A_thaliana, aligner:HISAT2])
[4e/aa282b] Submitted process > kangaAlign ([name:real, nreads:10k, seqerr:unk, rep:na, format:fq, ref:A_thaliana, aligner:BioKanga])
[99/cf76dd] Submitted process > extractStatsFromBAMs ([name:real, nreads:10k, seqerr:unk, rep:na, format:fq, ref:A_thaliana, aligner:HISAT2])
[27/a34b7f] Submitted process > multiQC
[24/858f7b] Submitted process > extractStatsFromBAMs ([name:real, nreads:10k, seqerr:unk, rep:na, format:fq, ref:A_thaliana, aligner:BioKanga])
[6b/c066be] Submitted process > combineStats
[23/3808f0] Submitted process > MOCK_generateFigures
[8a/610d6c] Submitted process > MOCK_generateReportMatterPipeline run (II)
N E X T F L O W ~ version 0.30.1
Launching `csiro-crop-informatics/reproducible_poc` [friendly_shockley] - revision: e957f4b275 [develop]
NOTE: Your local project version looks outdated - a different revision is available in the remote repository [5b495fe02f]
[warm up] executor > local
[12/5181e6] Cached process > fetchRef (A_thaliana)
[c2/8d5f02] Cached process > fetchReads
[e6/2bf4e9] Cached process > fastQC ([name:real, nreads:10k, seqerr:unk, rep:na, format:fq])
[ba/6e35da] Cached process > hisat2Index (A_thaliana)
[da/71e1b9] Cached process > kangaIndex (A_thaliana)
[29/8e7364] Cached process > hisat2Align ([name:real, nreads:10k, seqerr:unk, rep:na, format:fq, ref:A_thaliana, aligner:HISAT2])
[4e/aa282b] Cached process > kangaAlign ([name:real, nreads:10k, seqerr:unk, rep:na, format:fq, ref:A_thaliana, aligner:BioKanga])
[24/858f7b] Cached process > extractStatsFromBAMs ([name:real, nreads:10k, seqerr:unk, rep:na, format:fq, ref:A_thaliana, aligner:BioKanga])
[99/cf76dd] Cached process > extractStatsFromBAMs ([name:real, nreads:10k, seqerr:unk, rep:na, format:fq, ref:A_thaliana, aligner:HISAT2])
[05/5649ae] Submitted process > kangaSimReads ([name:A_thaliana, nreads:10000, seqerr:1.5, rep:1, format:fa])
[10/3aebcc] Submitted process > fasta2mockFASTQ ([name:A_thaliana, nreads:10000, seqerr:1.5, rep:1, format:fa])
[a0/ce8767] Submitted process > kangaAlign ([name:A_thaliana, nreads:10000, seqerr:1.5, rep:1, format:fa, ref:A_thaliana, aligner:BioKanga])
[71/62e3f7] Submitted process > hisat2Align ([name:A_thaliana, nreads:10000, seqerr:1.5, rep:1, format:fa, ref:A_thaliana, aligner:HISAT2])
[71/5b1259] Submitted process > fastQC ([name:A_thaliana, nreads:10000, seqerr:1.5, rep:1, format:fa])
[26/2bb088] Submitted process > extractStatsFromBAMs ([name:A_thaliana, nreads:10000, seqerr:1.5, rep:1, format:fa, ref:A_thaliana, aligner:BioKanga])
[1e/0f0bc9] Submitted process > extractStatsFromBAMs ([name:A_thaliana, nreads:10000, seqerr:1.5, rep:1, format:fa, ref:A_thaliana, aligner:HISAT2])
[0a/946feb] Submitted process > combineStats
[70/d0b8ad] Submitted process > MOCK_generateFigures
[36/4f3c57] Submitted process > multiQC
[e0/491389] Submitted process > MOCK_generateReportMatterPipeline flow
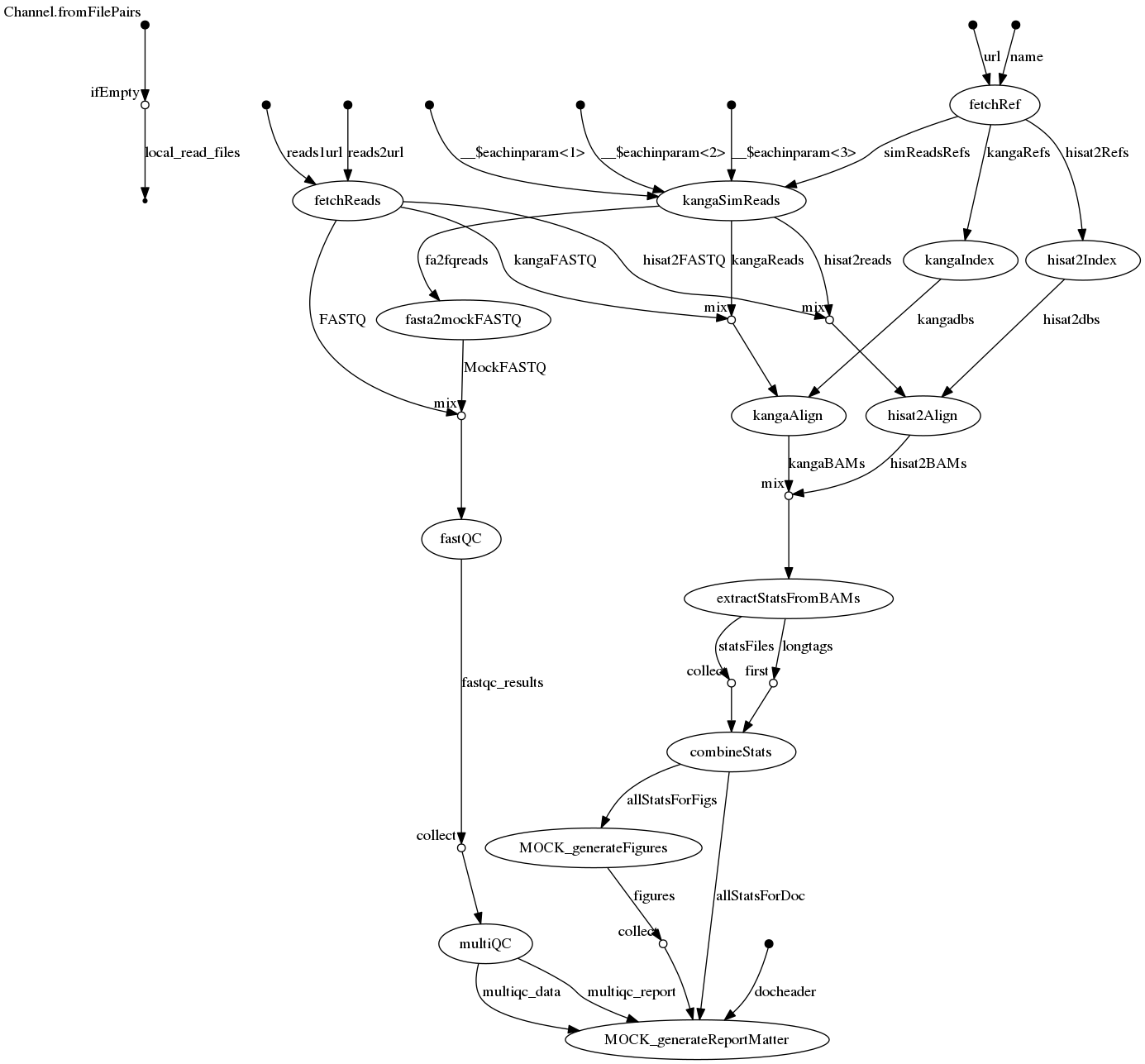
Pipeline syntax
- Primarily definitions of processes, each with well defined input and output values and/or files, usually passed through channels
//READ SIMULATION PARAMS
seqerrs = params.seqerrs.toString().tokenize(",")
nsimreadsarr = params.nsimreads.toString().tokenize(",")*.toInteger()
nrepeat = params.nrepeat
//INPUT GENOME PARAMS
url = params.url
name = params.name
//INPUT READS PARAMS
reads1url = params.realreads1
reads2url = params.realreads2
docheader = file(params.docheader)
def helpMessage() {
log.info"""
===========================================================
csiro-crop-informatics/reproducible_poc ~ version ${params.version}
===========================================================
Usage:
nextflow run csiro-crop-informatics/reproducible_poc -r develop
Default params:
seqerrs : ${params.seqerrs}
nsimreads : ${params.nsimreads} - this can be a comma-delimited list e.g. 100,20000,400
nrepeat : ${params.nrepeat}
url : ${params.url}
name : ${params.name}
outdir : ${params.outdir}
publishmode : ${params.publishmode} use copy or move if working across filesystems
""".stripIndent()
}
// Show help message
params.help = false
if (params.help){
helpMessage()
exit 0
}
/*
* Create a channel for (local) input read files
*/
//Channel
// .fromFilePairs( params.reads, size: 2 )
// .ifEmpty { exit 1, "Cannot find reads matching: ${params.reads}\nNB: Path must contain at least one * wildcard and be enclosed in quotes." }
// .set { local_read_files }
process fetchRef {
tag {name}
input:
val url
val name
output:
set val(name), file(ref) into kangaRefs, hisat2Refs, simReadsRefs
script:
"""
curl ${url} | gunzip --stdout | head -100000 > ref
"""
}
process fetchReads {
input:
val reads1url
val reads2url
output:
set val(longtag), val(nametag),file("r1.gz"), file("r2.gz") into FASTQ, hisat2FASTQ, kangaFASTQ
script:
nametag = "tmpTAG"
longtag = ["name":"real", "nreads":"10k", "seqerr":"unk", "rep":"na", "format":"fq"]
"""
curl ${reads1url} | gunzip --stdout | head -n 40000 | pigz --fast > r1.gz
curl ${reads2url} | gunzip --stdout | head -n 40000 | pigz --fast > r2.gz
"""
}
process kangaSimReads {
label 'biokanga'
tag {longtag}
input:
set val(name), file(ref) from simReadsRefs
each nsimreads from nsimreadsarr
each seqerr from seqerrs
each rep from 1..nrepeat
output:
set val(longtag), val(nametag),file("r1.gz"),file("r2.gz") into kangaReads, hisat2reads, fa2fqreads //simReads
when:
nsimreads > 0
script:
nametag = name+"_"+nsimreads+"_"+seqerr+"_"+rep
longtag = ["name":name, "nreads":nsimreads, "seqerr":seqerr, "rep":rep, "format":"fa"]
"""
biokanga simreads \
--pegen \
--seqerrs ${seqerr} \
--in ${ref} \
--nreads ${nsimreads} \
--out r1 \
--outpe r2 \
&& pigz --fast r1 r2
"""
}
process fasta2mockFASTQ {
tag {longtag}
input:
set val(longtag),val(nametag),file(r1),file(r2) from fa2fqreads
output:
set val(longtag), val(nametag), file ("*.q1.gz"), file("*.q2.gz") into MockFASTQ
"""
zcat ${r1} | fasta2fastqDummy.sh | pigz --fast --stdout > "${nametag}.q1.gz"
zcat ${r2} | fasta2fastqDummy.sh | pigz --fast --stdout > "${nametag}.q2.gz"
"""
}
process fastQC {
tag {longtag}
input:
set val(longtag), val(nametag), file("${nametag}.q1.gz"), file("${nametag}.q2.gz") from MockFASTQ.mix(FASTQ)
output:
file "*_fastqc.{zip,html}" into fastqc_results
"""
fastqc -q "${nametag}.q1.gz" "${nametag}.q2.gz"
"""
}
process multiQC {
input:
file f from fastqc_results.collect()
output:
file "*multiqc_report.html" into multiqc_report
file "*_data" into multiqc_data
"""
pwd
multiqc . -f
"""
}
process hisat2Index {
label 'hisat2'
tag{name}
input:
set val(name), file(ref) from hisat2Refs
output:
set val(name), file("hisat2db.*.ht2") into hisat2dbs
"""
hisat2-build ${ref} hisat2db -p 8
"""
}
process hisat2Align {
label 'hisat2'
label 'samtools'
tag {longtag}
input:
set val(longtag0), val(name), file(r1),file(r2) from hisat2reads.mix(hisat2FASTQ)
set val(dbname), file("hisat2db.*.ht2") from hisat2dbs
output:
set val(longtag), val(tag), file("${tag}.bam") into hisat2BAMs
script:
tag = name+"_vs_"+dbname+".hisat2"
longtag = longtag0.clone() //deepCopy(longtag0)
longtag.ref = dbname
longtag.aligner = "HISAT2"
format = longtag["format"]=="fq"?"-q":"-f"
"""
hisat2 -x hisat2db ${format} -1 ${r1} -2 ${r2} \
| samtools view -bS -F 4 -F 8 -F 256 - > ${tag}.bam
"""
}
process kangaIndex {
label 'biokanga'
tag{name}
input:
set val(name), file(ref) from kangaRefs
output:
set val(name), file(kangadb) into kangadbs
"""
biokanga index \
-i ${ref} \
-o kangadb \
--ref ${ref}
"""
}
process kangaAlign {
label 'biokanga'
tag {longtag}
input:
set val(longtag0), val(name),file(r1),file(r2) from kangaReads.mix(kangaFASTQ)
set val(dbname),file(kangadb) from kangadbs
output:
set val(longtag), val(tag), file("${tag}.bam") into kangaBAMs
script:
tag = name+"_vs_"+dbname+".biokanga"
longtag = longtag0.clone() //otherwise modifying orginal map, triggering re-runs with -resume
longtag.ref = dbname
longtag.aligner = "BioKanga"
"""
biokanga align \
-i ${r1} \
-u ${r2} \
--sfx ${kangadb} \
--threads ${task.cpus} \
-o "${tag}.bam" \
--pemode 2 \
"""
}
process extractStatsFromBAMs {
label 'samtools'
tag {longtag}
input:
set val(longtag), val(nametag), file("${nametag}*.bam") from kangaBAMs.mix(hisat2BAMs)
output:
file statsFile into statsFiles
val longtag into longtags
script:
statsPrefix = longtag.values().join("\t")+"\t"
"""
echo -ne "${statsPrefix}" > statsFile
samtools view ${nametag}.bam | extractStatsFromBAM.sh >> statsFile
"""
}
process combineStats {
input:
file("statsFile*") from statsFiles.collect()
val longtag from longtags.first()
output:
file allStats into allStatsForFigs, allStatsForDoc
script:
statsHeader = longtag.keySet().join("\t")+"\t"+"Matches\tAlignments\tMatchRate"
"""
cat <(echo -e "${statsHeader}") statsFile* >> allStats
"""
}
process MOCK_generateFigures {
label "MOCK_PROCESS"
input:
file allStats from allStatsForFigs
output:
file("*.figure") into figures
script:
"""
cat allStats > one.figure
cat allStats > another.figure
"""
// set file("*${nametag}.metadata"), file("*${nametag}.figure") into figures
// script:
// """
// echo "${nametag}" > "${nametag}.metadata"
// echo "${nametag}" > "${nametag}.figure"
// """
}
process MOCK_generateReportMatter {
label "MOCK_PROCESS"
input:
file "*figure" from figures.collect()
file allStats from allStatsForDoc
//set file(metadata), file(figure) from figures.collate(2)
//set val(nametag), file(statsFile) from statsFiles.collate(2)
file "*multiqc_report.html" from multiqc_report
file "*_data" from multiqc_data
file docheader
script:
"""
"""
// echo "---" > "${writeup}"
// cat "${docheader}" >> "${writeup}"
// echo -e "---\n" >> "${writeup}"
// echo "# Stats\n" >> "${writeup}"
// """
}
//process tagLocalReads {
// input:
// set val(name),file(reads) from local_read_files
//
// output:
// set val(longtag), val(nametag),file(r1), file(r2) into FASTQlocal //, hisat2FASTQlocal, kangaFASTQlocal
// exec:
// nametag = name
// longtag = ["name":"local", "nreads":"unk", "seqerr":"unk", "rep":"na", "format":"fq"]
//}These slides
- Source: nf-intro.Rmd
- Tools and packages:
- R, Pandoc,
rmarkdown,revealjs,kableExtra
- R, Pandoc,
- Use R, RStudio or a simple
RScriptto render from the command line
#!/usr/bin/env Rscript
args <- commandArgs(TRUE)
if(!require(rmarkdown)){
install.packages("rmarkdown")
library(rmarkdown)
}
if(!require(kableExtra)){
install.packages("kableExtra")
library(kableExtra)
}
if(!require(revealjs)){
location <- "~/local/R_libs/"; dir.create(location, recursive = TRUE)
install.packages("revealjs", lib=location, repos='https://cran.csiro.au')
library(revealjs, lib.loc=location)
}
rmarkdown::render(args[1], output_format = args[2])