Towards a reproducible and reusable publication and analysis workflow
Rad Suchecki
Nathan Watson-Haigh
Stuart Stephen
Alex Whan
Tuesday, 29 May, 2018
Reproducible manuscript - why?

- To avoid errors
- Widely reported inconsistencies between results and the methodology reported
- To promote computational reproducibility
- Other people (and you!) can take your data and get the same numbers that are in your paper
- Document must specify where ALL the numbers come from
- Otherwise numbers hard to recover even in absence of errors
- To create documents which can be revised easily
- New data, updated software or requests from reviewers can be incorporated much more easily
Ingredients for reproducibility in data science
- data
>1 dna:chromosome chromosome:TAIR10:1:1:30427671:1 REF
CCCTAAACCCTAAACCCTAAACCCTAAACCTCTGAATCCTTAATCCCTAAATCCCTAAATCTTTAAATCCTACATCCATGAATCCCTAAATACCTAATTCCCTAAACCCGAAACCGGTTTCTCTGGTTGAAAATCATTGTGTATATAATGATAATTTTATCGTTTTTATGTAATTGCTTA
TTGTTGTGTGTAGATTTTTTAAAAATATCATTTGAGGTCAATACAAATCCTATTTCTTGTGGTTTTCTTTCCTTCACTTAGCTATGGATGGTTTATCTTCATTTGTTATATTGGATACAAGCTTTGCTACGATCTACATTTGGGAATGTGAGTCTCTTATTGTAACCTTAGGGTTGGTTT
ATCTCAAGAATCTTATTAATTGTTTGGACTGTTTATGTTTGGACATTTATTGTCATTCTTACTCCTTTGTGGAAATGTTTGTTCTATCAATTTATCTTTTGTGGGAAAATTATTTAGTTGTAGGGATGAAGTCTTTCTTCGTTGTTGTTACGCTTGTCATCTCATCTCTCAATGATATGG- code
#!/bin/bash
awk -vOFS="\t" '{split($1,sim,"|");if(sim[4]==$3 && sim[5]==$4-1){count++}};END{print count,NR,count/NR}' ${@:-/dev/stdin}- compute environment
R version 3.4.4 (2018-03-15)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 16.04.4 LTS
Matrix products: default
BLAS: /usr/lib/libblas/libblas.so.3.6.0
LAPACK: /usr/lib/lapack/liblapack.so.3.6.0
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets base
other attached packages:
[1] revealjs_0.9 kableExtra_0.9.0 rmarkdown_1.9
loaded via a namespace (and not attached):
[1] Rcpp_0.12.17 rstudioapi_0.7 knitr_1.20
[4] xml2_1.2.0 magrittr_1.5 hms_0.4.2
[7] rvest_0.3.2 munsell_0.4.3 viridisLite_0.3.0
[10] colorspace_1.3-2 R6_2.2.2 rlang_0.2.0
[13] plyr_1.8.4 stringr_1.3.1 httr_1.3.1
[16] tools_3.4.4 htmltools_0.3.6 yaml_2.1.18
[19] rprojroot_1.3-2 digest_0.6.15 tibble_1.4.2
[22] readr_1.1.1 evaluate_0.10.1 stringi_1.2.2
[25] compiler_3.4.4 pillar_1.2.1 methods_3.4.4
[28] scales_0.5.0 backports_1.1.2 pkgconfig_2.0.1 BioKanga
- A suite of bioinformatics tools developed at CSIRO
- https://github.com/csiro-crop-informatics/biokanga
Aims
- evaluation of BioKanga’s sequence alignment module vs state-of-the-art tools
- turn-key reproducibility
- re-usability

Means
- open source and version control from day 1
- focus on design, not immediate result
- workflow management
- containers
Ongoing
- automated manuscript content generation
- tables, figures… not AI for writing text
Planned
- CI
- automated builds
Plans vs reality
- we have developed a modular workflow using
two popular automation frameworks, Snakemake andNextflow - Individual tasks are executed in Cloud or HPC environment using dedicated,
lightweight(Docker/Singularity) containers - Raw data is acquired from public sources, processed and analysed
- Final document including dynamically generated figures and tables is compiled from R Markdown
or LaTeX/knitr(minimal content at the moment, but works in principle)
Nextflow
A Workflow management system which promises
- Portability
- Scalability
- Reproducibility
Features
- Separation of pipeline logic from compute environment definition(s)
- Syntax is a superset of Groovy, but
- Polyglot
- mixing scripting languages, as long as interpreter available on system
- Multiple executors
- SGE, LSF, SLURM, PBS/Torque, NQSII HTCondor, Ignite, Kubernets, AWS Batch
Pipeline flow
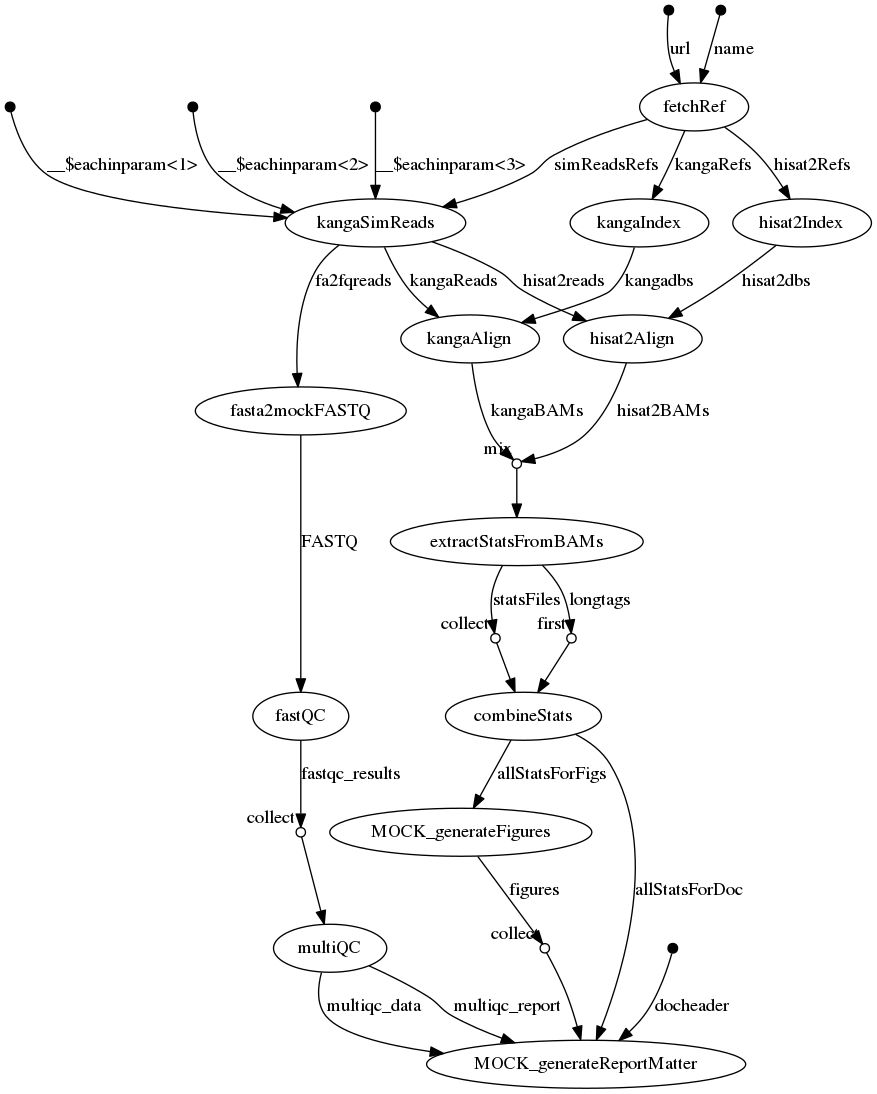
Pipeline syntax
- Primarily definitions of processes, each with well defined input and output channels
//READ SIMULATION PARAMS
seqerrs = params.seqerrs.toString().tokenize(",")
nreadsarr = params.nreads.toString().tokenize(",")
nrepeat = params.nrepeat
//INPUT GENOME PARAMS
url = params.url
name = params.name
docheader = file(params.docheader)
def helpMessage() {
log.info"""
===========================================================
csiro-crop-informatics/reproducible_poc ~ version ${params.version}
===========================================================
Usage:
nextflow run csiro-crop-informatics/reproducible_poc -r develop
Default params:
seqerrs : ${params.seqerrs}
nreads : ${params.nreads} - this can be a comma-delimited set e.g. 100,20000,400
nrepeat : ${params.nrepeat}
url : ${params.url}
name : ${params.name}
outdir : ${params.outdir}
publishmode : ${params.publishmode} use copy or move if working across filesystems
""".stripIndent()
}
// Show help message
params.help = false
if (params.help){
helpMessage()
exit 0
}
process fetchRef {
tag {name}
input:
val url
val name
output:
set val(name), file(ref) into kangaRefs, hisat2Refs, simReadsRefs
script:
"""
curl ${url} | gunzip --stdout > ref
"""
}
process kangaSimReads {
tag {longtag}
input:
set val(name), file(ref) from simReadsRefs
each nreads from nreadsarr
each seqerr from seqerrs
each rep from 1..nrepeat
output:
set val(longtag), val(nametag),file("r1.gz"),file("r2.gz") into kangaReads, hisat2reads, fa2fqreads //simReads
script:
nametag = name+"_"+nreads+"_"+seqerr+"_"+rep
longtag = ["name":name, "nreads":nreads, "seqerr":seqerr, "rep":rep]
"""
biokanga simreads \
--pegen \
--seqerrs ${seqerr} \
--in ${ref} \
--nreads ${nreads} \
--out r1 \
--outpe r2 \
&& pigz --fast r1 r2
"""
}
process fasta2mockFASTQ {
tag {longtag}
input:
set val(longtag),val(nametag),file(r1),file(r2) from fa2fqreads
output:
set val(longtag), val(nametag), file ("*.q1.gz"), file("*.q2.gz") into FASTQ
"""
zcat ${r1} | fasta2fastqDummy.sh | pigz --fast --stdout > "${nametag}.q1.gz"
zcat ${r2} | fasta2fastqDummy.sh | pigz --fast --stdout > "${nametag}.q2.gz"
"""
}
process fastQC {
tag {longtag}
input:
set val(longtag), val(nametag), file("${nametag}.q1.gz"), file("${nametag}.q2.gz") from FASTQ
output:
file "*_fastqc.{zip,html}" into fastqc_results
"""
fastqc -q "${nametag}.q1.gz" "${nametag}.q2.gz"
"""
}
process multiQC {
input:
file f from fastqc_results.collect()
output:
file "*multiqc_report.html" into multiqc_report
file "*_data" into multiqc_data
"""
pwd
multiqc . -f
"""
}
process hisat2Index {
tag{name}
input:
set val(name), file(ref) from hisat2Refs
output:
set val(name), file("hisat2db.*.ht2") into hisat2dbs
"""
hisat2-build ${ref} hisat2db -p 8
"""
}
process hisat2Align {
tag {longtag}
input:
set val(longtag0), val(name), file(r1),file(r2) from hisat2reads
set val(dbname), file("hisat2db.*.ht2") from hisat2dbs
output:
set val(longtag), val(tag), file("${tag}.bam") into hisat2BAMs
script:
tag = name+"_vs_"+dbname+".hisat2"
longtag = longtag0.clone() //deepCopy(longtag0)
longtag.ref = dbname
longtag.aligner = "HISAT2"
"""
hisat2 -x hisat2db -f -1 ${r1} -2 ${r2} \
| samtools view -bS -F 4 -F 8 -F 256 - > ${tag}.bam
"""
}
process kangaIndex {
tag{name}
input:
set val(name), file(ref) from kangaRefs
output:
set val(name), file(kangadb) into kangadbs
"""
biokanga index \
-i ${ref} \
-o kangadb \
--ref ${ref}
"""
}
process kangaAlign {
tag {longtag}
input:
set val(longtag0), val(name),file(r1),file(r2) from kangaReads
set val(dbname),file(kangadb) from kangadbs
output:
set val(longtag), val(tag), file("${tag}.bam") into kangaBAMs
script:
tag = name+"_vs_"+dbname+".biokanga"
longtag = longtag0.clone() //otherwise modifying orginal map, triggering re-runs with -resume
longtag.ref = dbname
longtag.aligner = "BioKanga"
"""
biokanga align \
-i ${r1} \
-u ${r2} \
--sfx ${kangadb} \
--threads ${task.cpus} \
-o "${tag}.bam" \
--pemode 2 \
"""
}
process extractStatsFromBAMs {
tag {longtag}
input:
set val(longtag), val(nametag), file("${nametag}*.bam") from kangaBAMs.mix(hisat2BAMs)
output:
file statsFile into statsFiles
val longtag into longtags
script:
statsPrefix = longtag.values().join("\t")+"\t"
"""
echo -ne "${statsPrefix}" > statsFile
samtools view ${nametag}.bam | extractStatsFromBAM.sh >> statsFile
"""
}
process combineStats {
input:
file("statsFile*") from statsFiles.collect()
val longtag from longtags.first()
output:
file allStats into allStatsForFigs, allStatsForDoc
script:
statsHeader = longtag.keySet().join("\t")+"\t"+"Matches\tAlignments\tMatchRate"
"""
cat <(echo -e "${statsHeader}") statsFile* >> allStats
"""
}
process MOCK_generateFigures {
label "MOCK_PROCESS"
input:
file allStats from allStatsForFigs
output:
file("*.figure") into figures
script:
"""
cat allStats > one.figure
cat allStats > another.figure
"""
// set file("*${nametag}.metadata"), file("*${nametag}.figure") into figures
// script:
// """
// echo "${nametag}" > "${nametag}.metadata"
// echo "${nametag}" > "${nametag}.figure"
// """
}
process MOCK_generateReportMatter {
label "MOCK_PROCESS"
input:
file "*figure" from figures.collect()
file allStats from allStatsForDoc
//set file(metadata), file(figure) from figures.collate(2)
//set val(nametag), file(statsFile) from statsFiles.collate(2)
file "*multiqc_report.html" from multiqc_report
file "*_data" from multiqc_data
file docheader
script:
"""
"""
// echo "---" > "${writeup}"
// cat "${docheader}" >> "${writeup}"
// echo -e "---\n" >> "${writeup}"
// echo "# Stats\n" >> "${writeup}"
// """
}Compute profiles
Implemented
standard(i.e. local/interactive assuming all software available)slurmmodulesdockersingularityec2
# some of the profiles can, and others are intended to be used in concert, e.g.
nextflow run main.nf -profile slurm,modules
# or
nextflow run main.nf -profile slurm,singularity,singularitymoduleUpcoming:
k8s,awsbatch
Modules
- Required modules are defined separately and explicitly for each process (or associated with labels)
process {
$fastQC.module = 'fastqc/0.11.5'
$multiQC.module = 'python/2.7.13'
$kangaSimReads.module = 'biokanga/4.3.9'
$kangaIndex.module = 'biokanga/4.3.9'
$kangaAlign.module = 'biokanga/4.3.9'
$hisat2Index.module = 'hisat/2.0.5'
$hisat2Align.module = ['hisat/2.0.5', 'samtools/1.7.0']
$extractStatsFromBAMs.module = 'samtools/1.7.0'
}- Unfortunately, these are compute environment specific
Containers
- Required containers are defined separately and explicitly for each process (or associated with labels)
- Available on docker hub
- Used both by
dockerandsingularity - Always specify version tag (avoid
':latest')
- Used both by
process {
$fastQC.container = 'genomicpariscentre/fastqc:0.11.5'
$multiQC.container = 'ewels/multiqc:v1.5'
$fasta2mockFASTQ.container = 'rsuchecki/pigz:2.3.4'
$kangaSimReads.container = 'rsuchecki/biokanga:4.3.9'
$kangaIndex.container = 'rsuchecki/biokanga:4.3.9'
$kangaAlign.container = 'rsuchecki/biokanga:4.3.9'
$hisat2Index.container = 'cyverse/hisat2:2.0.5' //also contains samtools
$hisat2Align.container = 'cyverse/hisat2:2.0.5' //also contains samtools
$extractStatsFromBAMs.container = 'genomicpariscentre/samtools:1.4.1'
}Generating reports/publications/slides from R Markdown

Some of the supported output formats:
- HTML* documents and slide-shows using
- Slidy, ioslides, reveal.js
- PDF
- LaTeX
- LaTeX Beamer slides
- MS Word, ODT, RTF
- Markdown, and Github flavoured Markdown documents
These slides
- Source: show.Rmd
- Tools and packages:
- R, Pandoc,
rmarkdown,revealjs,kableExtra
- R, Pandoc,
- Use R, RStudio or a simple
RScriptto render from the command line
#!/usr/bin/env Rscript
args <- commandArgs(TRUE)
if(!require(rmarkdown)){
install.packages("rmarkdown")
library(rmarkdown)
}
if(!require(kableExtra)){
install.packages("kableExtra")
library(kableExtra)
}
if(!require(revealjs)){
location <- "~/local/R_libs/"; dir.create(location, recursive = TRUE)
install.packages("revealjs", lib=location, repos='https://cran.csiro.au')
library(revealjs, lib.loc=location)
}
rmarkdown::render(args[1], output_format = args[2])Report generation
Design conundrum
Pipeline to render the documentDocument rendering to execute pipeline- Run pipeline, then render the document
Detail
- output from the tested tools evaluated against ground truth
- speed benchmarking
Workflow introspection
Tables and figures dynamically generated from collected results
dt <- read.table("results/stats/allStats", header=TRUE, sep="\t")
#kable(dt[with(dt, order(MatchRate, decreasing=TRUE)), ]) %>%
kable(dt) %>%
scroll_box(height = "400px") #add width for horizontal scroll, can use % as well| name | nreads | seqerr | rep | ref | aligner | Matches | Alignments | MatchRate |
|---|---|---|---|---|---|---|---|---|
| A_thaliana | 10000 | 1 | 1 | A_thaliana | BioKanga | 19048 | 19048 | 1.00000 |
| A_thaliana | 10000 | 3 | 2 | A_thaliana | HISAT2 | 19689 | 20000 | 0.98445 |
| A_thaliana | 10000 | 3 | 2 | A_thaliana | BioKanga | 19012 | 19012 | 1.00000 |
| A_thaliana | 10000 | 2 | 2 | A_thaliana | HISAT2 | 19690 | 20000 | 0.98450 |
| A_thaliana | 10000 | 1 | 3 | A_thaliana | HISAT2 | 19656 | 20000 | 0.98280 |
| A_thaliana | 10000 | 1 | 3 | A_thaliana | BioKanga | 18936 | 18936 | 1.00000 |
| A_thaliana | 10000 | 2 | 3 | A_thaliana | HISAT2 | 19656 | 20000 | 0.98280 |
| A_thaliana | 10000 | 2 | 3 | A_thaliana | BioKanga | 18936 | 18936 | 1.00000 |
| A_thaliana | 10000 | 1 | 4 | A_thaliana | BioKanga | 19076 | 19076 | 1.00000 |
| A_thaliana | 10000 | 1 | 4 | A_thaliana | HISAT2 | 19708 | 20000 | 0.98540 |
| A_thaliana | 10000 | 3 | 3 | A_thaliana | HISAT2 | 19653 | 20000 | 0.98265 |
| A_thaliana | 10000 | 3 | 1 | A_thaliana | BioKanga | 19116 | 19116 | 1.00000 |
| A_thaliana | 10000 | 3 | 3 | A_thaliana | BioKanga | 18948 | 18948 | 1.00000 |
| A_thaliana | 10000 | 2 | 4 | A_thaliana | HISAT2 | 19677 | 20000 | 0.98385 |
| A_thaliana | 10000 | 2 | 4 | A_thaliana | BioKanga | 18994 | 18994 | 1.00000 |
| A_thaliana | 10000 | 1 | 5 | A_thaliana | HISAT2 | 19677 | 20000 | 0.98385 |
| A_thaliana | 10000 | 1 | 5 | A_thaliana | BioKanga | 18994 | 18994 | 1.00000 |
| A_thaliana | 10000 | 3 | 4 | A_thaliana | HISAT2 | 19687 | 20000 | 0.98435 |
| A_thaliana | 10000 | 3 | 4 | A_thaliana | BioKanga | 18974 | 18974 | 1.00000 |
| A_thaliana | 10000 | 2 | 5 | A_thaliana | HISAT2 | 19676 | 20000 | 0.98380 |
| A_thaliana | 10000 | 2 | 5 | A_thaliana | BioKanga | 19046 | 19046 | 1.00000 |
| A_thaliana | 10000 | 3 | 5 | A_thaliana | HISAT2 | 19694 | 20000 | 0.98470 |
| A_thaliana | 10000 | 1 | 1 | A_thaliana | HISAT2 | 19693 | 20000 | 0.98465 |
| A_thaliana | 10000 | 3 | 5 | A_thaliana | BioKanga | 19074 | 19074 | 1.00000 |
| A_thaliana | 10000 | 3 | 1 | A_thaliana | HISAT2 | 19711 | 20000 | 0.98555 |
| A_thaliana | 10000 | 1 | 2 | A_thaliana | BioKanga | 19116 | 19116 | 1.00000 |
| A_thaliana | 10000 | 2 | 2 | A_thaliana | BioKanga | 19050 | 19050 | 1.00000 |
| A_thaliana | 10000 | 2 | 1 | A_thaliana | BioKanga | 19048 | 19048 | 1.00000 |
| A_thaliana | 10000 | 2 | 1 | A_thaliana | HISAT2 | 19693 | 20000 | 0.98465 |
| A_thaliana | 10000 | 1 | 2 | A_thaliana | HISAT2 | 19711 | 20000 | 0.98555 |
Further Work (I)
Document generation refinement
- R Markdown rendering not as flexible as hoped
- Generation of output in different formats somewhat limited, for more complex content where pure (R)Markdown is insufficient have to decide on either
- (R)Markdown with LaTeX
- (R)Markdown with HTML
Workflow
- Comparison of Nextflow and Snakemake (limited, in context)
- Tagged containers for
- each release of the pipeline
- submitted/revised/final publication
Further Work (II)
- More systematic evaluation of BioKanga alignment module vs other tools
- At default settings
- Exploration of parameter space, optimisation
- More alignment tools to be included
(Some of) the good
- https://gitlab.com/makhlaghi/reproducible-paper (
makeandLaTeX) - https://github.com/Robinlovelace/rmarkdown-paper-repo
- https://github.com/libscie/rmarkdown-workshop
- https://andrewgoldstone.com/blog/2015/05/27/rmd-slides/
The bad & the ugly
4 pages of this. Images pasted into a word document! pic.twitter.com/CkVpLutZZV
— Paul Gardner (@ppgardne) May 27, 2018